Spatial Domain Identification#
Spatial transcriptomics data from bin-based platforms like Visium captures gene expression across tissue sections without single-cell resolution, where each spot may contain multiple cells. Identifying spatial domains — contiguous tissue regions with distinct molecular signatures — is a key analytical goal that reveals tissue architecture and microenvironment organization. This notebook demonstrates spatial domain identification using the exprmat package with multiple complementary algorithms. We cover spatially variable gene detection with SpatialDE, domain detection using SpaGCN with and without histological image integration, and deep learning-based domain identification with DeepST. We also demonstrate integration of spatial domains across multiple samples and the identification of domain-specific marker genes.
[1]:
%load_ext autoreload
%autoreload 2
[2]:
import exprmat as em
em.setwd('../../../data')
ver = em.version()
[i] exprmat 0.2.66 / exprmat-db 0.2.66
[i] os: posix (linux) platform version: 6.8.0-90-generic
[i] loaded configuration from /home/data/yangz/.exprmatrc
[i] current working directory: /home/data/yangz/packages/exprmat/data
[i] current database directory: /home/data/yangz/packages/database (0.2.66)
[i] resident memory: 777.58 MiB
[i] virtual memory: 5.95 GiB
[3]:
expm = em.load_experiment('expm/visium')
━━━━━━━━━━━━━━━━━━━━━━━━━━ loading samples 8 / 8 (00:06 < 00:00)
Identification of spatial domains is commonly useful in bin-based data. Since this gives an abstraction of cellular neighborhood in spatial coordinates. For single cell resolution expression matrices, a direct approach of cellular neighbors binning after cell type identification is more accurate and meaningful, (see tutorial d1 and e1) though the same algorithm still works, it is not a recommended workflow.
[6]:
print(expm)
annotated data of size 12038 × 35554
integrated dataset of size 12038 × 35554
contains modalities: spatial-bin
modality [spatial-bin]
obs : sample <cat> batch <cat> group <cat> modality <cat> taxa <cat> barcode <o> ubc <o>
in.tissue <i64> row <i64> col <i64> y <i64> x <i64>
var : chr <cat> start <i64> end <i64> strand <cat> id <o> subtype <cat> gene <cat> tlen <f64>
cdslen <i64> assembly <cat> uid <o>
obsm : spatial <arr:i64(2)>
uns : spatial
[*] composed of samples:
p1-t4 spatial-bin hsa batch b1 of size 1155 × 35554
p1-t5 spatial-bin hsa batch b2 of size 1726 × 35554
p1-t7 spatial-bin hsa batch b3 of size 1191 × 35554
p1-t10 spatial-bin hsa batch b4 of size 1522 × 35554
p2-t1 spatial-bin hsa batch b5 of size 2197 × 35554
p2-t2 spatial-bin hsa batch b6 of size 1853 × 35554
p2-t5 spatial-bin hsa batch b7 of size 1470 × 35554
p2-t6 spatial-bin hsa batch b8 of size 924 × 35554
SpatialDE#
Identification of spatially variable genes is often performed sample-wise. Before merged to the whole dataset. SpatialDE requires counts layer input
[5]:
sample = 'p2-t5'
[8]:
expm.spatial_bin.view(sample)
annotated data of size 1470 × 35554
obs : sample <cat> batch <cat> group <cat> modality <cat> taxa <cat> barcode <o> ubc <o>
in.tissue <i64> row <i64> col <i64> y <i64> x <i64> leiden <cat>
var : chr <cat> start <i64> end <i64> strand <cat> id <o> subtype <cat> gene <cat> tlen <f64>
cdslen <i64> assembly <cat> uid <o> vst.means <f64> vst.vars <f64> vst.vars.norm <f64>
vst.hvg.rank <f32> vst.hvg <bool>
layers : counts <f32> lognorm <f32> norm <f32>
obsm : knn <arr:i32(30)> knn.d <arr:f64(30)> pca <arr:f64(35)> spatial <arr:i64(2)>
spatial.array <arr:i64(2)> umap <arr:f32(2)>
varm : pca <arr:f64(35)>
obsp : connectivities <csr:f32> distances <csr:f64>
uns : leiden neighbors pca spatial umap
[9]:
expm.spatial_bin.spatial_svg_spatialde(
run_on_samples = [sample],
layer = 'counts',
spatial_key = 'spatial',
kernel_space = None,
sizefactors = None,
obs_dist = 'negative_binomial',
use_cache = True,
use_gpu = True,
progress = True,
n_svg = 500,
key_added = 'spatialde'
)
[i] performing spatialde score test for svgs ...
━━━━━━━━━━━━━━━━━━━━━━━━━━ spatialde score test 35554 / 35554 (05:13 < 00:00)
[i] tested 35554 genes; 1845 significant at fdr 5%.
[9]:
{'p2-t5': None}
[15]:
expm.spatial_bin[sample].var[['gene', 'spatialde.padj', 'spatialde.svg', 'spatialde.rank']] \
.sort_values('spatialde.rank').head(15)
[15]:
| gene | spatialde.padj | spatialde.svg | spatialde.rank | |
|---|---|---|---|---|
| rna:hsa:g38601 | CSTB | 5.124839e-11 | True | 1.0 |
| rna:hsa:g11501 | HSPA8 | 5.124839e-11 | True | 2.0 |
| rna:hsa:g52277 | PERP | 5.124839e-11 | True | 3.0 |
| rna:hsa:g57815 | PABPC1 | 5.124839e-11 | True | 4.0 |
| rna:hsa:g57829 | YWHAZ | 5.124839e-11 | True | 5.0 |
| rna:hsa:g31642 | CMPK2 | 5.124839e-11 | True | 6.0 |
| rna:hsa:g31643 | RSAD2 | 5.124839e-11 | True | 7.0 |
| rna:hsa:g16460 | COL4A1 | 5.124839e-11 | True | 8.0 |
| rna:hsa:g19674 | DUOX2 | 5.124839e-11 | True | 9.0 |
| rna:hsa:g19675 | DUOXA2 | 5.124839e-11 | True | 10.0 |
| rna:hsa:g19696 | C15orf48 | 5.124839e-11 | True | 11.0 |
| rna:hsa:g19654 | B2M | 5.124839e-11 | True | 12.0 |
| rna:hsa:g45142 | RPL34 | 5.124839e-11 | True | 13.0 |
| rna:hsa:g44891 | SPP1 | 5.124839e-11 | True | 14.0 |
| rna:hsa:g38502 | MX2 | 5.124839e-11 | True | 15.0 |
[28]:
fig = expm.spatial_bin.plot_spatial(
run_on_samples = [sample],
channels = ['hires/r', 'hires/g', 'hires/b'],
plot_embeddings = {
'visible': True,
'basis': 'spatial',
'color': 'CSTB',
'ptsize': 15,
},
xrange = (3000, 13000),
yrange = (5000, 15000),
figsize = (5, 5)
)
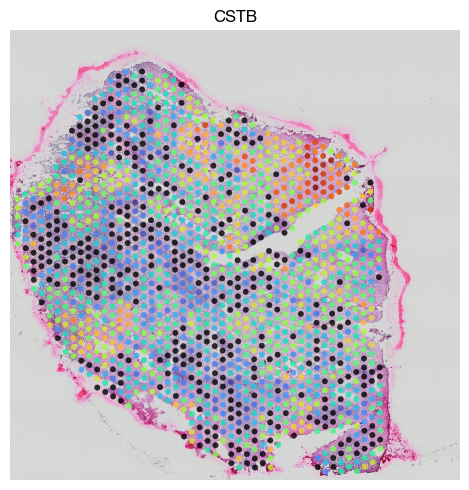
Such spatially variable gene can be intersected to highly variable genes to perform HVG selection and later dimensionality reduction process with spatially-awared properties
Spatial domain identification#
Several methods are available to identify spatial domains with similar expression profile beforehand, and then infer gene signature of those domains. These offer visually explainable gene signatures with direct mapping to tissue distribution.
SpaGCN provides a robust implementation of such algorithms. It requires a normalized expression matrix to fit the model. User should have a estimated number of clusters and may run the method several time to achieve requested resolution. An H&E image may also be provided for image embeddings
[37]:
expm.spatial_bin.spatial_detect_domains_spagcn(
run_on_samples = [sample],
layer = 'lognorm',
channels = None,
n_clusters = 7,
histology = False, # at present without histology images.
refine_shape = 'hexagon',
seed = 42,
key_added = 'spagcn.domain',
refined_key_added = 'spagcn.refined'
)
[i] calculating adjacency matrix from xy coordinates only.
[i] recommended l = 90.829404296875
[i] search_res starting at res = 0.70, step = 0.10.
[i] res = 0.60, n_clusters = 8.
[i] delta_label 0.004762 < tol 0.005, stopping at epoch 19.
[i] res = 0.50, n_clusters = 5.
[i] res = 0.55, n_clusters = 8.
[i] delta_label 0.004762 < tol 0.005, stopping at epoch 19.
[i] res = 0.50, n_clusters = 5.
[i] res = 0.52, n_clusters = 8.
[i] delta_label 0.004762 < tol 0.005, stopping at epoch 19.
[i] res = 0.50, n_clusters = 5.
[i] res = 0.51, n_clusters = 8.
[i] delta_label 0.004762 < tol 0.005, stopping at epoch 19.
[i] res = 0.50, n_clusters = 5.
[i] res = 0.51, n_clusters = 7.
[i] delta_label 0.004762 < tol 0.005, stopping at epoch 22.
[i] calculating adjacency matrix from xy coordinates only.
[31]:
fig = expm.spatial_bin.plot_spatial(
run_on_samples = [sample],
channels = ['hires/r', 'hires/g', 'hires/b'],
plot_embeddings = {
'visible': True,
'basis': 'spatial',
'color': 'spagcn.domain',
'ptsize': 15,
},
xrange = (3000, 13000),
yrange = (5000, 15000),
figsize = (5, 5)
)
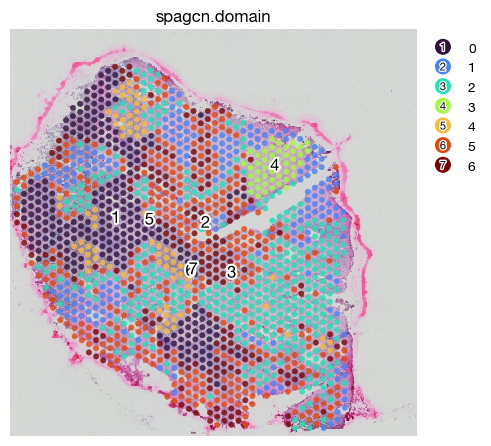
[32]:
fig = expm.spatial_bin.plot_spatial(
run_on_samples = [sample],
channels = ['hires/r', 'hires/g', 'hires/b'],
plot_embeddings = {
'visible': True,
'basis': 'spatial',
'color': 'spagcn.refined',
'ptsize': 15,
},
xrange = (3000, 13000),
yrange = (5000, 15000),
figsize = (5, 5)
)
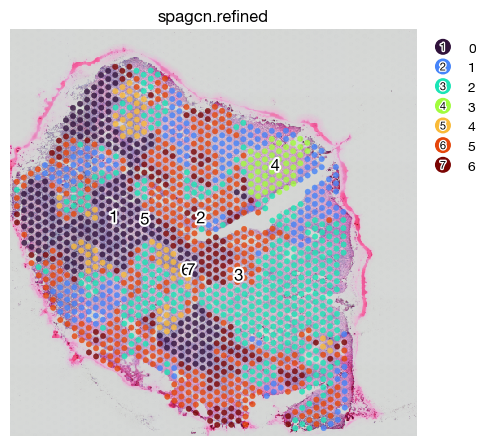
You may also retrieve the high-resolution tissue image to faciliate tissue-morphology based spatial domain identification
[39]:
he = expm.spatial_bin.get_image(
run_on_samples = ['p1-t4'],
channels = ['hires/r', 'hires/g', 'hires/b']
)
[36]:
expm.spatial_bin.spatial_detect_domains_spagcn(
run_on_samples = [sample],
layer = 'lognorm',
channels = ['hires/r', 'hires/g', 'hires/b'],
n_clusters = 7,
histology = True,
refine_shape = 'hexagon',
seed = 42,
key_added = 'spagcn.domain',
refined_key_added = 'spagcn.refined'
)
[i] calculating adjacency matrix from spatial coords + histology image.
[i] recommended l = 121.1025390625
[i] search_res starting at res = 0.70, step = 0.10.
[i] res = 0.60, n_clusters = 8.
[i] res = 0.50, n_clusters = 6.
[i] res = 0.55, n_clusters = 7.
[i] delta_label 0.004082 < tol 0.005, stopping at epoch 43.
[i] calculating adjacency matrix from xy coordinates only.
[34]:
fig = expm.spatial_bin.plot_spatial(
run_on_samples = [sample],
channels = ['hires/r', 'hires/g', 'hires/b'],
plot_embeddings = {
'visible': True,
'basis': 'spatial',
'color': 'spagcn.refined',
'ptsize': 15,
},
xrange = (3000, 13000),
yrange = (5000, 15000),
figsize = (5, 5)
)
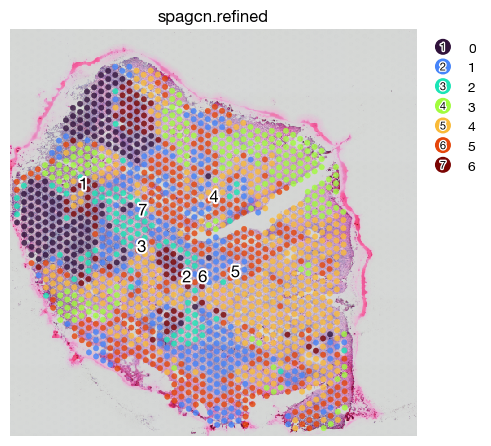
Spatial domain identification using DeepST#
DeepST provides an alternative to SpaGCN. Though the result is less convincing in this case.
[38]:
expm.spatial_bin.spatial_detect_domains_deepst(
run_on_samples = [sample],
image_key = 'hires',
n_domains = 7,
use_image = False,
save_path = './expm/deepst',
pre_epochs = 200,
epochs = 200,
use_gpu = True
)
[i] spatial weights computed; average neighbours = 30.0.
[i] gene-expression weights computed.
[i] combined weight matrix saved to adata.obsm['weights'].
━━━━━━━━━━━━━━━━━━━━━━━━ finding adjacent spots 1470 / 1470 (00:01 < 00:00)
[i] data augmentation completed.
[i] 12.0000 neighbours per cell on average.
[i] spatial graph computation completed.
[i] running deepst training ...
━━━━━━━━━━━━━━━━━━━━━ pretraining initial model 200 / 200 (00:05 < 00:00)
━━━━━━━━━━━━━━━━━━━━━━━━━━ training final model 200 / 200 (00:05 < 00:00)
[i] deepst training finished. memory usage = 4.58 GB; total time = 0.20 min.
[i] found resolution 0.41 for 7 domains.
[i] deepst domain detection complete.
[39]:
expm.spatial_bin.view(sample)
annotated data of size 1470 × 35554
obs : sample <cat> batch <cat> group <cat> modality <cat> taxa <cat> barcode <o> ubc <o>
in.tissue <i64> row <i64> col <i64> y <i64> x <i64> leiden <cat> spagcn.domain <cat>
spagcn.refined <cat> imagerow <f64> imagecol <f64> deepst.domain <cat>
deepst.refined <cat>
var : chr <cat> start <i64> end <i64> strand <cat> id <o> subtype <cat> gene <cat> tlen <f64>
cdslen <i64> assembly <cat> uid <o> vst.means <f64> vst.vars <f64> vst.vars.norm <f64>
vst.hvg.rank <f32> vst.hvg <bool> spatialde.padj <f64> spatialde.svg <bool>
spatialde.rank <f64>
layers : counts <f32> lognorm <f32> norm <f32>
obsm : knn <arr:i32(30)> knn.d <arr:f64(30)> pca <arr:f64(35)> spatial <arr:i64(2)>
spatial.array <arr:i64(2)> umap <arr:f32(2)> weights <arr:f64> adjacent <arr:f64(35554)>
augmented.genes <arr:f64(35554)> deepst.embedding <arr:f32(28)>
varm : pca <arr:f64(35)>
obsp : connectivities <csr:f32> distances <csr:f64>
uns : leiden neighbors pca spatial umap deepst.domain
[40]:
fig = expm.spatial_bin.plot_spatial(
run_on_samples = [sample],
channels = ['hires/r', 'hires/g', 'hires/b'],
plot_embeddings = {
'visible': True,
'basis': 'spatial',
'color': 'deepst.refined',
'ptsize': 15,
},
xrange = (3000, 13000),
yrange = (5000, 15000),
figsize = (5, 5)
)
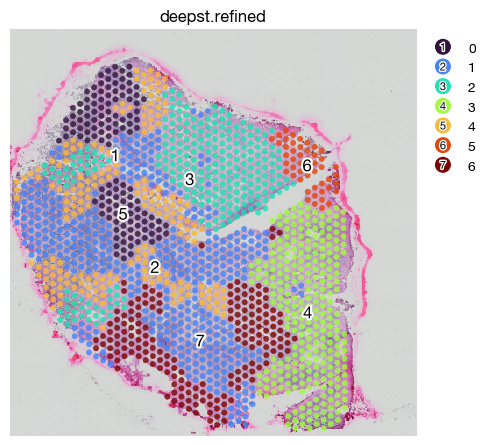
I do not recommend running DeepST on data other than large bins, since the intermediate storage of per-spot image clips may occupy much storage for cells greater than 5000.
[41]:
expm.spatial_bin.spatial_detect_domains_deepst(
run_on_samples = [sample],
image_key = 'hires',
n_domains = 7,
use_image = True,
save_path = './expm/deepst',
pre_epochs = 3000,
epochs = 3000,
use_gpu = True
)
━━━━━━━━━━━━━━━━━━━━━━━━━━ cropping spot images 1470 / 1470 (00:38 < 00:00)
━━━━━━━━━━━━━━━━━━━━━ extracting image features 1470 / 1470 (00:17 < 00:00)
[i] spatial weights computed; average neighbours = 30.0.
[i] gene-expression weights computed.
[i] morphological weights computed.
[i] combined weight matrix saved to adata.obsm['weights'].
━━━━━━━━━━━━━━━━━━━━━━━━ finding adjacent spots 1470 / 1470 (00:01 < 00:00)
[i] data augmentation completed.
[i] 12.0000 neighbours per cell on average.
[i] spatial graph computation completed.
[i] running deepst training ...
━━━━━━━━━━━━━━━━━━━━━ pretraining initial model 3000 / 3000 (01:15 < 00:00)
━━━━━━━━━━━━━━━━━━━━━━━━━━ training final model 3000 / 3000 (01:29 < 00:00)
[i] deepst training finished. memory usage = 5.21 GB; total time = 2.75 min.
[!] no resolution gave exactly 7 domains; falling back to resolution = 0.2.
[i] deepst domain detection complete.
[42]:
fig = expm.spatial_bin.plot_spatial(
run_on_samples = [sample],
channels = ['hires/r', 'hires/g', 'hires/b'],
plot_embeddings = {
'visible': True,
'basis': 'spatial',
'color': 'deepst.refined',
'ptsize': 15,
},
xrange = (3000, 13000),
yrange = (5000, 15000),
figsize = (5, 5)
)
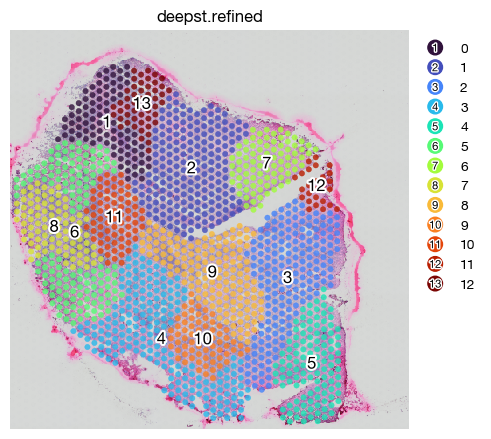
Integrating multiple datasets to uniformed domains#
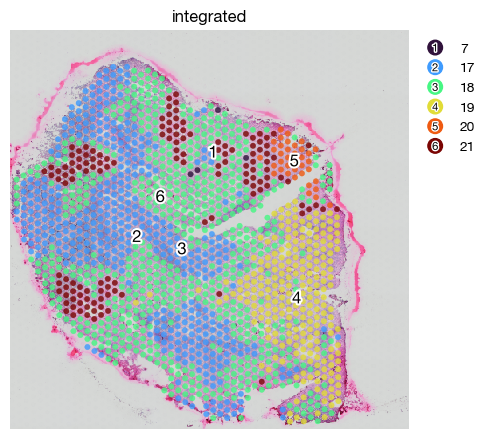
Spatial integration are often useful to identify patterns shared across different samples. This generalizability provides an easy access to comparisons. Since DeepST did not perform well on tumor tissues, we prefer SpaGCN integration of domains.
[ ]:
expm.spatial_bin.integrate_domains_spagcn(
n_clusters = 15,
run_on_samples = True, # integrate all samples.
result_key = 'integrated',
histology = False,
max_epochs = 500,
seed = 100,
)
[i] delta_label 0.004901 < tol 0.005, stopping at epoch 70.
[45]:
fig = expm.spatial_bin.plot_spatial(
run_on_samples = [sample],
channels = ['hires/r', 'hires/g', 'hires/b'],
plot_embeddings = {
'visible': True,
'basis': 'spatial',
'color': 'integrated',
'ptsize': 15,
},
xrange = (3000, 13000),
yrange = (5000, 15000),
figsize = (5, 5)
)
Identify domain-specific markers#
Like we obtain marker from unsupervised clustering, such spatial domains can use specially designed methods for marker gene retrieval
[ ]:
expm.spatial_bin.spatial_markers_spagcn(
run_on_samples = [sample],
domain_key = 'integrated',
min_in_group_fraction = 0.8,
min_in_out_group_ratio = 1,
min_fold_change = 1.5,
num_neighbours = 3,
ratio = 0.5,
radius_quantiles = (0.001, 0.1),
num_min = 10,
num_max = 14,
max_run = 100,
key_added = 'spagcn.markers'
)
[ ]:
expm.spatial_bin.get_markers(
run_on_samples = [sample],
slot = 'spagcn.markers',
group_name = '17'
)[sample][['gene', 'pct.in', 'pct.out', 'fc', 'q', 'log10.q']]
[i] fetched diff `17` over `rest` (18 genes)
| gene | pct.in | pct.out | fc | q | log10.q | |
|---|---|---|---|---|---|---|
| rna:hsa:g13826 | LUM | 1.000000 | 0.996835 | 2.302415 | 6.437318e-78 | 77.191295 |
| rna:hsa:g22861 | HERPUD1 | 0.997691 | 0.995253 | 2.107135 | 6.339839e-77 | 76.197922 |
| rna:hsa:g53680 | SFRP4 | 0.852194 | 0.485759 | 2.073682 | 2.236636e-110 | 109.650405 |
| rna:hsa:g15444 | POSTN | 0.946882 | 0.783228 | 1.945651 | 3.066666e-95 | 94.513333 |
| rna:hsa:g57703 | TP53INP1 | 0.997691 | 0.992089 | 1.802479 | 7.859763e-72 | 71.104591 |
| rna:hsa:g59915 | ASPN | 0.967667 | 0.799051 | 1.755815 | 1.035827e-91 | 90.984713 |
| rna:hsa:g57902 | CTHRC1 | 0.963048 | 0.806962 | 1.665744 | 3.148795e-84 | 83.501856 |
| rna:hsa:g6228 | VIM | 1.000000 | 0.995253 | 1.655484 | 1.246817e-58 | 57.904197 |
| rna:hsa:g4692 | BTG2 | 0.997691 | 0.995253 | 1.605542 | 1.204427e-58 | 57.919219 |
| rna:hsa:g62036 | PIM2 | 0.995381 | 0.985759 | 1.579871 | 1.728512e-58 | 57.762328 |
| rna:hsa:g45757 | SFRP2 | 0.933025 | 0.912975 | 1.579464 | 7.928045e-74 | 73.100834 |
| rna:hsa:g35422 | IGFBP5 | 0.956120 | 0.943038 | 1.578879 | 2.130125e-68 | 67.671595 |
| rna:hsa:g58420 | PTP4A3 | 1.000000 | 0.985759 | 1.565828 | 1.001160e-74 | 73.999496 |
| rna:hsa:g39969 | FBLN1 | 0.935335 | 0.881329 | 1.562546 | 6.522984e-76 | 75.185554 |
| rna:hsa:g52137 | CCN2 | 0.995381 | 0.988924 | 1.557651 | 3.224739e-44 | 43.491505 |
| rna:hsa:g35923 | COL6A3 | 0.990762 | 0.973101 | 1.552273 | 5.541407e-72 | 71.256380 |
| rna:hsa:g44744 | CXCL13 | 0.956120 | 0.941456 | 1.518375 | 1.463188e-57 | 56.834700 |
| rna:hsa:g34966 | COL5A2 | 0.997691 | 0.977848 | 1.515412 | 1.668277e-65 | 64.777732 |
[57]:
fig = expm.spatial_bin.plot_spatial(
run_on_samples = [sample],
channels = ['hires/r', 'hires/g', 'hires/b'],
plot_embeddings = {
'visible': True,
'basis': 'spatial',
'color': 'LUM',
'ptsize': 15,
},
xrange = (3000, 13000),
yrange = (5000, 15000),
figsize = (5, 5)
)

Detect metagenes#
SpaGCN provides a simple linear model of gene combinations to introduce high specificity marker set as metagenes
[11]:
expm.spatial_bin.spatial_metagene(
run_on_samples = [sample],
domain_key = 'integrated',
target_cluster = '17',
key_svg = 'spagcn.markers',
max_iter = 6,
key_added = 'metagene.17'
)
[i] add gene: rna:hsa:g18771; minus gene: rna:hsa:g3509
[i] absolute mean change: 4.9681
[i] non-target spots reduced to: 149
[i] meta gene now: rna:hsa:g58420 + rna:hsa:g18771 - rna:hsa:g3509
[i] add gene: rna:hsa:g39127; minus gene: rna:hsa:g53115
[i] absolute mean change: 7.4248
[i] non-target spots reduced to: 10
[i] meta gene now: rna:hsa:g58420 + rna:hsa:g18771 - rna:hsa:g3509 + rna:hsa:g39127 - rna:hsa:g53115
[i] meta gene: rna:hsa:g58420 + rna:hsa:g18771 - rna:hsa:g3509 + rna:hsa:g39127 - rna:hsa:g53115
[i] meta gene: rna:hsa:g58420 + rna:hsa:g18771 - rna:hsa:g3509 + rna:hsa:g39127 - rna:hsa:g53115
[13]:
fig = expm.spatial_bin.plot_spatial(
run_on_samples = [sample],
channels = ['hires/r', 'hires/g', 'hires/b'],
plot_embeddings = {
'visible': True,
'basis': 'spatial',
'color': 'metagene.17',
'ptsize': 15,
},
xrange = (3000, 13000),
yrange = (5000, 15000),
figsize = (5, 5)
)

[17]:
expm.spatial_bin.view(sample)
annotated data of size 1470 × 35554
obs : sample <cat> batch <cat> group <cat> modality <cat> taxa <cat> barcode <o> ubc <o>
in.tissue <i64> row <i64> col <i64> y <i64> x <i64> leiden <cat> spagcn.domain <cat>
spagcn.refined <cat> imagerow <f64> imagecol <f64> deepst.domain <cat>
deepst.refined <cat> integrated <cat> metagene.17 <f64>
var : chr <cat> start <i64> end <i64> strand <cat> id <o> subtype <cat> gene <cat> tlen <f64>
cdslen <i64> assembly <cat> uid <o> vst.means <f64> vst.vars <f64> vst.vars.norm <f64>
vst.hvg.rank <f32> vst.hvg <bool> spatialde.padj <f64> spatialde.svg <bool>
spatialde.rank <f64>
layers : counts <f32> lognorm <f32> norm <f32>
obsm : adjacent <arr:f64(35554)> augmented.genes <arr:f64(35554)> deepst.embedding <arr:f32(28)>
knn <arr:i32(30)> knn.d <arr:f64(30)> pca <arr:f64(35)> spatial <arr:i64(2)>
spatial.array <arr:i64(2)> umap <arr:f32(2)> weights <arr:f64>
varm : pca <arr:f64(35)>
obsp : connectivities <csr:f32> distances <csr:f64>
uns : deepst.domain leiden neighbors pca spagcn.markers spatial umap
[18]:
expm.save()
[i] main dataset write to expm/visium/integrated.h5mu
[i] saving individual samples. (pass `save_samples = False` to skip)
━━━━━━━━━━━━━━━━━━━━━━━━ modality [spatial-bin] 8 / 8 (00:07 < 00:00)