Statistics#
We introduce several basic utility functions for summarizing and computing statistics on sparse matrices.
[1]:
%load_ext autoreload
%autoreload 2
[2]:
import exprmat as em
# set working directory
em.setwd('../../../data')
ver = em.version()
[i] exprmat 0.2.66 / exprmat-db 0.2.66
[i] os: posix (linux) platform version: 6.8.0-90-generic
[i] loaded configuration from /home/data/yangz/.exprmatrc
[i] current working directory: /home/data/yangz/packages/exprmat/data
[i] current database directory: /home/data/yangz/packages/database (0.2.66)
[i] resident memory: 778.73 MiB
[i] virtual memory: 5.95 GiB
[3]:
expm = em.load_experiment('expm/scrna', load_subset = 'mono-neutro')
━━━━━━━━━━━━━━━━━━━━━━━━━━ loading samples 3 / 3 (00:03 < 00:00)
[4]:
print(expm)
annotated data of size 9754 × 19651
subset mono-neutro of size 9754 × 19651
contains modalities: rna
modality [rna]
obs : sample <cat> <c/sample> batch <cat> <c/batch> group <cat> <c> modality <cat> <c/modality>
taxa <cat> <c/taxa> barcode <o> <o> ubc <o> <o> n.umi <f64> <i> n.genes <i64> <i>
n.mito <f64> <f> n.ribo <f64> <f> pct.mito <f64> <f> pct.ribo <f64> <f>
filter <bool> <bool> score.doublet <f64> <f> score.doublet.se <f64> <f>
is.doublet <bool> <bool> qc <bool> <bool/qc> leiden <cat> <c> sc3.5 <cat> <c>
sc3.10 <cat> <c> sc3.20 <cat> <c> sc3.30 <cat> <c> cell.type <cat> <c>
var : chr <cat> <c/chromosome> start <i64> <i> end <i64> <i> strand <cat> <c/strand> id <o> <o>
subtype <cat> <c/gsubtype> gene <cat> <o/gene> tlen <f64> <i/tlen> cdslen <i64> <i/cdslen>
assembly <cat> <c> uid <o> <o/ugene> vst.hvg <bool> <bool/hvg> vst.all.means <f64> <f>
vst.all.vars <f64> <f> vst.all.vars.norm <f64> <f> vst.all.hvg.rank <f32> <f>
vst.all.hvg <bool> <bool>
layers : counts <f32> <i/counts> norm <f32> <f>
obsm : cnmf.10 <df> <f/embedding/usage> harmony <arr:f32(35)> <f> knn <arr:i32(100)> <i/knni>
knn.d <arr:f32(100)> <f/knnd> pca <arr:f64(35)> <f/embedding/pca>
umap <arr:f32(2)> <f/embedding>
varm : cnmf.10 <arr:f64(10)> <f/weights> cnmf.coef.10 <arr:f64(10)> <f/usage-coef>
pca <arr:f64(35)> <f/weights>
obsp : connectivities <csr:f32> <f/connectivity> distances <csr:f32> <f/distance>
uns : cell.type.colors cell.type_colors cnmf <cnmf> cnmf.args <o>
cnmf.density.10 <cnmf-density> cnmf.dist.10 <f/connectivity> cnmf.stats <cnmf-stats>
commands <system> leiden <o> leiden.colors <o> markers <markers> neighbors <knn>
pca <dict> sc3.10.colors <o> sc3.20.colors <o> sc3.30.colors <o> sc3.5.colors <o>
slots <system> umap <o>
[*] composed of samples:
distal rna mmu batch b1 of size 6451 × 21689
niche rna mmu batch b2 of size 3656 × 21970
normal rna mmu batch b3 of size 4537 × 21917
Cluster proportion statistics#
Compute the proportions of categorical variables across dataset rows (cells). The major parameter specifies the primary grouping variable, while minor specifies the categorical variable for which proportions are calculated.
[7]:
expm.rna.proportion(
major = 'sample', minor = 'cell.type',
normalize = 'index'
)['integrated']
[7]:
| cell.type | DCp | MDP | MM | Mo | Neu | Prog | iMac |
|---|---|---|---|---|---|---|---|
| sample | |||||||
| distal | 0.023297 | 0.062052 | 0.080421 | 0.035842 | 0.774194 | 0.023970 | 0.000224 |
| niche | 0.001878 | 0.160345 | 0.079985 | 0.088246 | 0.563275 | 0.024784 | 0.081487 |
| normal | 0.065855 | 0.159878 | 0.051389 | 0.031976 | 0.666159 | 0.023601 | 0.001142 |
Gaussian kernel density estimation#
Given a low-dimensional representation, we can compute the density of the scatter point distribution to visualize how cells are concentrated in different regions of the embedding space.
[8]:
fig = expm.rna.plot_embedding(
basis = 'umap', color = 'cell.type',
figsize = (3, 3), dpi = 100, legend_col = 2, legend = False,
ptsize = 2, annotate_style = 'text', annotate_fontsize = 8, contour_plot = True
)
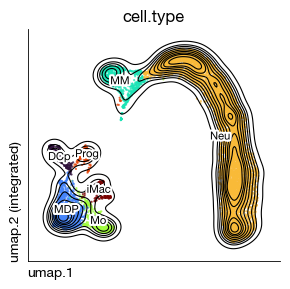
[9]:
expm.rna.kde(
basis = "umap",
groupby = "sample",
key_added = "kde.umap",
components = "1,2"
)
[10]:
kde = expm.rna.plot_kde(
basis = 'umap', kde = 'kde.umap', grouping_key = 'sample',
background = 'cell.type', annotate = True, annotate_fontsize = 8, ptsize = 2,
figsize = (9, 3), dpi = 100, groups = None, ncols = 3, cmap = 'turbo'
)
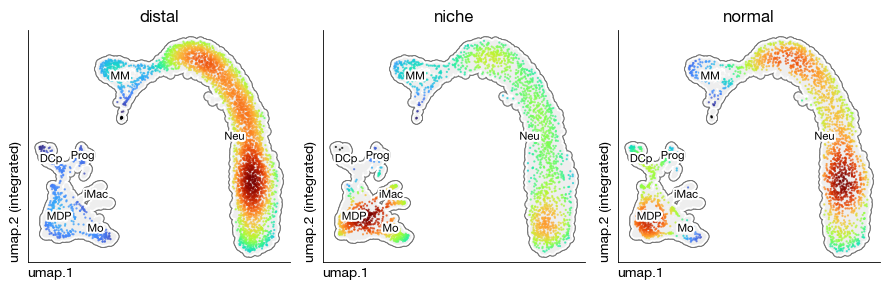
Summary statistics#
The summary function creates a three-dimensional table using the on, across, and split parameters. Each cell value in the table is computed from the data, orient, and method parameters. The returned object is an AnnData structure where rows correspond to on, columns correspond to across, and layers contain each split.
When orient is set to 'obs', the on, across, and split parameters must be column names in the obs table. Similarly, when orient is set to 'var', these must be column names in the var table. Specific rows or columns are selected by the three filter conditions, and the method function is applied to produce the cell values of the three-dimensional table. If the filtered result is an empty set, the corresponding cell is NaN.
When method is set to the special method n, it returns the count of observations (rows or columns, not the count of values within the statistical range).
[13]:
adata = expm.rna.summary(
data = 'X', method = 'n', method_args = {},
orient = 'obs', on = 'sample',
across = 'cell.type', split = 'sample',
attached_metadata_on = None, attached_metadata_across = None,
attach_method_on = 'first', attach_method_across = 'first'
)['integrated']
[14]:
em.pd.DataFrame(
adata.layers['niche'],
index = adata.obs_names,
columns = adata.var_names
)
[14]:
| Neu | MDP | MM | Mo | DCp | Prog | iMac | |
|---|---|---|---|---|---|---|---|
| distal | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| niche | 1500.0 | 427.0 | 213.0 | 235.0 | 5.0 | 66.0 | 217.0 |
| normal | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
Aggregate statistics#
The aggregate function collapses the original data table along rows or columns, applying a specified method (such as sum or mean) to groups defined by observation or variable keys.
[15]:
adata = expm.rna.aggregate(
data = 'counts', method = 'sum', method_args = {},
obs_key = 'sample', var_key = None
)['integrated']
[16]:
em.pd.DataFrame(
adata.X,
index = adata.obs_names,
columns = adata.var['gene']
).iloc[:, 0:10]
[16]:
| gene | Bzw1 | Ppp1r9b | Samd14 | Clk1 | Pdk2 | Itga3 | Ppil3 | Tac4 | Kat7 | Gm11520 |
|---|---|---|---|---|---|---|---|---|---|---|
| distal | 4153.0 | 1685.0 | 29.0 | 6723.0 | 54.0 | 0.0 | 456.0 | 2.0 | 1604.0 | 144.0 |
| niche | 3908.0 | 2584.0 | 42.0 | 4752.0 | 46.0 | 11.0 | 442.0 | 6.0 | 1415.0 | 110.0 |
| normal | 3470.0 | 1561.0 | 21.0 | 4831.0 | 72.0 | 1.0 | 446.0 | 8.0 | 1292.0 | 129.0 |
Pseudobulking#
Pseudobulking merges cells from a specified category by counts, and continue the analysis normalization as if they were coming from bulk dataset.
[17]:
expm.rna.annotate_paste(
froms = ['sample', 'cell.type'],
into = 'psbulk',
slot = 'rna',
sep = ':'
)
psbulk
distal:Neu 3456
normal:Neu 1750
niche:Neu 1500
niche:MDP 427
normal:MDP 420
distal:MM 359
distal:MDP 277
niche:Mo 235
niche:iMac 217
niche:MM 213
normal:DCp 173
distal:Mo 160
normal:MM 135
distal:Prog 107
distal:DCp 104
normal:Mo 84
niche:Prog 66
normal:Prog 62
niche:DCp 5
normal:iMac 3
distal:iMac 1
Name: count, dtype: int64
[19]:
expm.rna.pseudobulk(
obs_key = 'psbulk',
sample_added = 'psbulk',
obs_metadata = ['cell.type', 'sample', 'group'],
merge_obs_metadata = 'mode'
)
[i] created subset [psbulk] from sample [integrated]
[20]:
print(expm)
annotated data of size 9754 × 19651
subset mono-neutro of size 9754 × 19651
contains modalities: rna
modality [rna]
obs : sample <cat> <c/sample> batch <cat> <c/batch> group <cat> <c> modality <cat> <c/modality>
taxa <cat> <c/taxa> barcode <o> <o> ubc <o> <o> n.umi <f64> <i> n.genes <i64> <i>
n.mito <f64> <f> n.ribo <f64> <f> pct.mito <f64> <f> pct.ribo <f64> <f>
filter <bool> <bool> score.doublet <f64> <f> score.doublet.se <f64> <f>
is.doublet <bool> <bool> qc <bool> <bool/qc> leiden <cat> <c> sc3.5 <cat> <c>
sc3.10 <cat> <c> sc3.20 <cat> <c> sc3.30 <cat> <c> cell.type <cat> <c>
kde.umap <f64> <f/kde> psbulk <cat>
var : chr <cat> <c/chromosome> start <i64> <i> end <i64> <i> strand <cat> <c/strand> id <o> <o>
subtype <cat> <c/gsubtype> gene <cat> <o/gene> tlen <f64> <i/tlen> cdslen <i64> <i/cdslen>
assembly <cat> <c> uid <o> <o/ugene> vst.hvg <bool> <bool/hvg> vst.all.means <f64> <f>
vst.all.vars <f64> <f> vst.all.vars.norm <f64> <f> vst.all.hvg.rank <f32> <f>
vst.all.hvg <bool> <bool>
layers : counts <f32> <i/counts> norm <f32> <f>
obsm : cnmf.10 <df> <f/embedding/usage> harmony <arr:f32(35)> <f> knn <arr:i32(100)> <i/knni>
knn.d <arr:f32(100)> <f/knnd> pca <arr:f64(35)> <f/embedding/pca>
umap <arr:f32(2)> <f/embedding>
varm : cnmf.10 <arr:f64(10)> <f/weights> cnmf.coef.10 <arr:f64(10)> <f/usage-coef>
pca <arr:f64(35)> <f/weights>
obsp : connectivities <csr:f32> <f/connectivity> distances <csr:f32> <f/distance>
uns : cell.type.colors cell.type_colors cnmf <cnmf> cnmf.args <o>
cnmf.density.10 <cnmf-density> cnmf.dist.10 <f/connectivity> cnmf.stats <cnmf-stats>
commands <system> leiden <o> leiden.colors <o> markers <markers> neighbors <knn>
pca <dict> sc3.10.colors <o> sc3.20.colors <o> sc3.30.colors <o> sc3.5.colors <o>
slots <system> umap <o> kde.umap <kde-stats>
[*] composed of samples:
distal rna mmu batch b1 of size 6451 × 21689
niche rna mmu batch b2 of size 3656 × 21970
normal rna mmu batch b3 of size 4537 × 21917
psbulk rna autogen batch autogen of size 21 × 19651
[21]:
fig = expm.rna.plot_pseudobulk_samples(
run_on_samples = ['psbulk'],
groupby = None, figsize = (3, 3),
min_cells = 10, min_counts = 1000,
log = True,
)
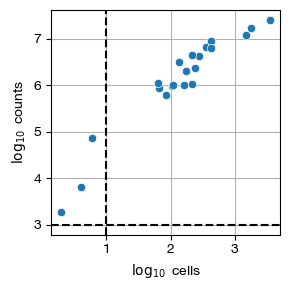
[22]:
expm.rna.filter_pseudobulk_samples(
run_on_samples = ['psbulk'],
min_cells = 10, min_counts = 1000,
)
[23]:
expm.rna.view('psbulk')
annotated data of size 18 × 19651
obs : cell.type <o> <c> sample <o> <c/sample> group <o> <c> counts <f32> <f> cells <i64> <i>
var : chr <cat> <c/chromosome> start <i64> <i> end <i64> <i> strand <cat> <c/strand> id <o> <o>
subtype <cat> <c/gsubtype> gene <cat> <o/gene> tlen <f64> <i/tlen> cdslen <i64> <i/cdslen>
assembly <cat> <c> uid <o> <o/ugene> vst.hvg <bool> <bool/hvg> vst.all.means <f64> <f>
vst.all.vars <f64> <f> vst.all.vars.norm <f64> <f> vst.all.hvg.rank <f32> <f>
vst.all.hvg <bool> <bool>
layers : proportions <f64> <f/proportions>
uns : slots <system> commands <system>
[24]:
fig = expm.rna.plot_pseudobulk_features(
run_on_samples = ['psbulk'],
group = 'group',
lib_size = None,
min_count = 10,
min_total_count = 15,
large_n = 10,
min_sample_prop = 0.7,
cmap = 'viridis',
min_cell_prop = 0.2,
min_samples = 2,
figsize = (7, 3),
dpi = 100
)
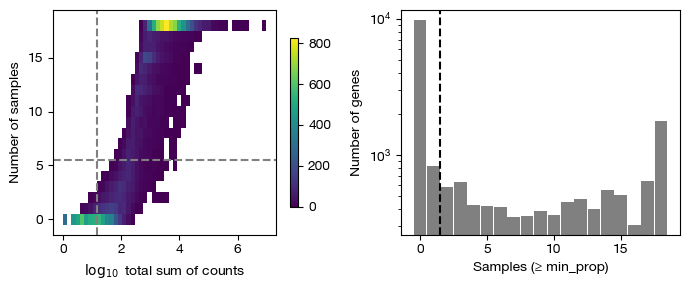
[25]:
expm.rna.filter_pseudobulk_features(
run_on_samples = ['psbulk'],
group = 'group',
lib_size = None,
min_count = 10,
min_total_count = 15,
large_n = 10,
min_sample_prop = 0.7,
min_cell_prop = 0.2,
min_samples = 2
)
[27]:
expm.save()
[i] main dataset write to expm/scrna/subsets/mono-neutro.h5mu
[i] saving individual samples. (pass `save_samples = False` to skip)
━━━━━━━━━━━━━━━━━━━━━━━━━━ modality [rna] 4 / 4 (00:01 < 00:00)